PRO-seq T4 RNA ligase correction and analysis
We will explore the sequence bias assocated with single nucleotide resolution GRO-seq (Core et al., 2008): PRO-seq (Kwak et al., 2013). All PRO-seq data is from K562 cells (Core et al., 2014).
Retrieving and processing PRO-seq data
mkdir Core_PRO
wget ftp://ftp-trace.ncbi.nlm.nih.gov/sra/sra-instant/reads/ByExp/sra/SRX/SRX683/SRX683602/SRR1554311/SRR1554311.sra
fastq-dump SRR1554311.sra
mv SRR1554311.fastq K562_pro.fastq
rm SRR1554311.sra
fastx_clipper -Q 33 -i K562_pro.fastq -o K562_pro.clipped.fastq -a TGGAATTCTCGGGTGCCAAGG -l 15
rm K562_pro.fastq
fastx_trimmer -Q 33 -l 30 -i K562_pro.clipped.fastq -o K562_pro.trimmed.fastq
rm K562_pro.clipped.fastq
fastx_reverse_complement -Q 33 -z -i K562_pro.trimmed.fastq -o K562_pro.rc.fastq.gz
rm K562_pro.trimmed.fastq
bowtie2 -x ~/DNase_ENCODE/hg38 -U K562_pro.rc.fastq.gz -S K562_pro.sam
samtools view -b K562_pro.sam | samtools sort - K562_pro
rm K562_pro.sam
#process plus and minus aligned reads separately
samtools view -bh -F 20 K562_pro.bam > K562_pro_plus.bam
samtools view -bh -f 0x10 K562_pro.bam > K562_pro_minus.bam
Using seqOutBias to correct PRO-seq data
We will not perform genomic k-mer correction, instead we will will look exclusively at gene annotations. The vast majority of transcription occurs in annotated genes, although lower levels of transcription are pervasive in the genome. The reason we are looking at genes is because it is conceivable that the k-mer counts are different between the genome and the transcribed units.
#gene annotations
wget ftp://ftp.ensembl.org/pub/release-87/gtf/homo_sapiens/Homo_sapiens.GRCh38.87.gtf.gz
gunzip gunzip Homo_sapiens.GRCh38.87.gtf.gz
awk '$3 == "gene"' Homo_sapiens.GRCh38.87.gtf | sed 's/^/chr/' | awk '{OFS="\t";} {print $1,$4,$5,$2,$6,$7}' > Homo_sapiens.GRCh38.87.bed
awk '$3 == "gene"' Homo_sapiens.GRCh38.87.gtf | sed 's/^/chr/' | awk '{OFS="\t";} {print $1,$4,$5,$2,$6,$7}' > Homo_sapiens.GRCh38.87.bed
awk '$6 == "+"' Homo_sapiens.GRCh38.87.bed | awk '{OFS="\t";} {print $1,$2,$2+100,$4,$5,$6}' > Homo_sapiens.GRCh38.87.plus.dsTSS.bed
awk '$6 == "-"' Homo_sapiens.GRCh38.87.bed | awk '{OFS="\t";} {print $1,$3-100,$3,$4,$5,$6}' > Homo_sapiens.GRCh38.87.minus.dsTSS.bed
cat Homo_sapiens.GRCh38.87.plus.dsTSS.bed Homo_sapiens.GRCh38.87.minus.dsTSS.bed > Homo_sapiens.GRCh38.87.dsTSS.bed
subtractBed -s -a Homo_sapiens.GRCh38.87.bed -b Homo_sapiens.GRCh38.87.dsTSS.bed > Homo_sapiens.GRCh38.87.body.bed
awk '$6 == "+"' Homo_sapiens.GRCh38.87.body.bed > Homo_sapiens.GRCh38.87.body.plus.bed
awk '$6 == "-"' Homo_sapiens.GRCh38.87.body.bed > Homo_sapiens.GRCh38.87.body.minus.bed
sort -k1,1 -k2,2n Homo_sapiens.GRCh38.87.body.plus.bed > Homo_sapiens.GRCh38.87.body.plus.sorted.bed
sort -k1,1 -k2,2n Homo_sapiens.GRCh38.87.body.minus.bed > Homo_sapiens.GRCh38.87.body.minus.sorted.bed
rm Homo_sapiens.GRCh38.87.body.plus.bed
rm Homo_sapiens.GRCh38.87.body.minus.bed
mergeBed -s -i Homo_sapiens.GRCh38.87.body.plus.sorted.bed > Homo_sapiens.GRCh38.87.body.plus.bed
mergeBed -s -i Homo_sapiens.GRCh38.87.body.minus.sorted.bed > Homo_sapiens.GRCh38.87.body.minus.bed
We will process the reads that align to the plus and minus strand separately and implement the tail-edge option to output the 3’ end of the sequence read.
#correct based on k-mers observed in gene bodies
reg=Homo_sapiens.GRCh38.87.body
bam=K562_pro_plus.bam
seqOutBias ~/DNase_ENCODE/hg38.fa $bam --regions=${reg}.plus.bed --no-scale --bw=PRO_plus_body_0-mer.bigWig --tail-edge --read-size=30
bam=K562_pro_minus.bam
seqOutBias ~/DNase_ENCODE/hg38.fa $bam --regions=${reg}.minus.bed --no-scale --bw=PRO_minus_body_0-mer.bigWig --tail-edge --read-size=30
bam=K562_pro_plus.bam
seqOutBias ~/DNase_ENCODE/hg38.fa $bam --regions=${reg}.plus.bed --kmer-mask=NNNCNNN --bw=PRO_plus_body_NNNCNNN-mer.bigWig --tail-edge --read-size=30
bam=K562_pro_minus.bam
seqOutBias ~/DNase_ENCODE/hg38.fa $bam --regions=${reg}.minus.bed --kmer-mask=NNNCNNN --bw=PRO_minus_body_NNNCNNN-mer.bigWig --tail-edge --read-size=30
#for loading into UCSC
for bw in *plus*-mer.bigWig
do
name=$(echo $bw | awk -F"/" '{print $NF}' | awk -F".bigWig" '{print $1}')
echo $name
bigWigToBedGraph $bw $name.bg
touch temp.txt
echo "track type=bedGraph name=$name color=255,0,0 alwaysZero=on visibility=full" >> temp.txt
cat temp.txt $name.bg > $name.bedGraph
rm temp.txt
rm $name.bg
gzip $name.bedGraph
done
for bw in *minus*mer.bigWig
do
name=$(echo $bw | awk -F"/" '{print $NF}' | awk -F".bigWig" '{print $1}')
echo $name
bigWigToBedGraph $bw $name.bg
touch temp.txt
echo "track type=bedGraph name=$name color=0,0,255 alwaysZero=on visibility=full" >> temp.txt
cat temp.txt $name.bg > $name.bedGraph
rm temp.txt
rm $name.bg
gzip $name.bedGraph
done
mkdir plus
mkdir minus
mv *minus*bigWig minus
mv *plus*bigWig plus
Plotting PRO-seq density surrounding TF binding sites
Next we will look at PRO-seq signal centered around CTCF binding sites. We will consider the orientation of the CTCF motif and the original alignment strand of the sequence read.
wget http://hgdownload.cse.ucsc.edu/goldenPath/hg19/encodeDCC/wgEncodeUwTfbs/wgEncodeUwTfbsK562CtcfStdPkRep1.narrowPeak.gz
for peak in *narrowPeak.gz
do
name=$(echo $peak | awk -F"wgEncodeUwTfbsK562" '{print $NF}' | awk -F"sc" '{print $1}' | awk -F"Std" '{print $1}')
unz=$(echo $peak | awk -F".gz" '{print $1}')
echo $name
gunzip $peak
echo $unz
liftOver $unz ~/DNase_ENCODE/hg19ToHg38.over.chain $name.hg38.narrowPeak $name.hg38.narrow.unmapped.txt -bedPlus=6
fastaFromBed -fi ~/DNase_ENCODE/hg38.fa -bed $name.hg38.narrowPeak -fo $name.hg38.fasta
gzip *narrowPeak
done
#specifically find peaks in the gene bodies of genes on the + and - strands #specifically look at CTCF binding sites in both orientations
mv Ctcf.hg38.fasta CTCF.hg38.fasta
mv Ctcf.hg38.narrowPeak.gz CTCF.hg38.k562.narrowPeak.gz
head -9 ~/DNase_ENCODE/motif_databases/JASPAR/JASPAR_CORE_2016_vertebrates.meme > header_meme_temp.txt
grep -i -A 23 'MOTIF MA0139.1 CTCF' ~/DNase_ENCODE/motif_databases/JASPAR/JASPAR_CORE_2016.meme > ctcf_temp.txt
cat header_meme_temp.txt ctcf_temp.txt > CTCF_minimal_meme.txt
rm *temp.txt
gunzip CTCF.hg38.k562.narrowPeak
intersectBed -a CTCF.hg38.k562.narrowPeak -b Homo_sapiens.GRCh38.87.body.plus.bed > CTCF.hg38.k562.gb.plus.peaks.bed
fastaFromBed -fi ~/DNase_ENCODE/hg38.fa -bed CTCF.hg38.k562.gb.plus.peaks.bed -fo CTCF.hg38.k562.gb.plus.peaks.fasta
intersectBed -a CTCF.hg38.k562.narrowPeak -b Homo_sapiens.GRCh38.87.body.minus.bed > CTCF.hg38.k562.gb.minus.peaks.bed
fastaFromBed -fi ~/DNase_ENCODE/hg38.fa -bed CTCF.hg38.k562.gb.minus.peaks.bed -fo CTCF.hg38.k562.gb.minus.peaks.fasta
for meme in *.peaks.fasta
do
name=$(echo $meme | awk -F".hg38" '{print $1}') nm=$(echo $meme | awk -F".peaks" '{print $1}')
echo $name
mast ${name}_minimal_meme.txt $meme -hit_list -mt 0.0005 > ${nm}_mast.txt
done
for i in *_mast.txt
do
name=$(echo $i | awk -F"_mast" '{print $1}')
grep ' +1 ' $i > ${name}_plus_mast.txt
grep ' -1 ' $i > ${name}_minus_mast.txt
done
source('https://raw.githubusercontent.com/guertinlab/seqOutBias/master/docs/R/seqOutBias_functions.R')
all.composites.plus.pro = cycle.fimo.new.not.hotspots(path.dir.mast = '~/Core_PRO/',
path.dir.bigWig = '/Users/guertinlab/Core_PRO/plus/', window = 30, exp = 'PRO plus')
all.composites.minus.pro = cycle.fimo.new.not.hotspots(path.dir.mast = '~/Core_PRO/',
path.dir.bigWig = '/Users/guertinlab/Core_PRO/minus/', window = 30, exp = 'PRO minus')
save(all.composites.minus.pro, all.composites.plus.pro, file = 'PRO_composites.Rdata')
composites.func.pro(all.composites.plus.pro, fact= "PRO plus", summit= "CTCF motif",num =24)
composites.func.pro(all.composites.minus.pro, fact= "PRO minus", summit= "CTCF motif",num =24)
Plotting PRO-seq density surrounding splice sites
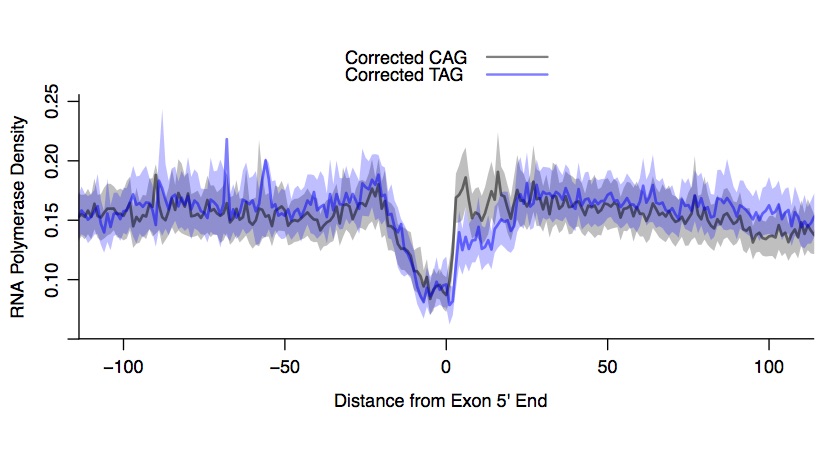
Previous work in Drosophila has shown that RNA Polymerase density decreases directly upstream of the 5’ end of exons, at the site of the 3’ splice site (Kwak et al., 2013). To determine whether the run on experiment or the library preparation exhibit sequence biases, we plot the PRO-seq density at the 5’ exon boundary. We will exclude the first exon from our analysis, because RNA Polymerase II pausing is a common feature of most genes. We will process the plus and minus strand genes separately. Additionally, we will plot the composites for distinct splice acceptor sequences. Positions -3 relative to the exon start tolerates all nucleotides, but C and T are prefered. As expected, the span of the confidence intervals correlates with the number of motif instances in each category (Figure 8). Therefore, we randomly selected 38358 CAG rows to match the 38358 TAG rows–there are many fewer instances of the AAG 3’ splice acceptor, so we excluded these. The composite profiles show that the CAG consensus splice site (compared to TAG) promotes slower elongation rate in the 5 ́end of exons (Figure 9).
awk '$3 == "exon"' Homo_sapiens.GRCh38.87.gtf | sed 's/^/chr/' | grep -v 'exon_number "1"' | awk '{OFS="\t";} {print $1,$4,$5,$2,$6,$7}' > Homo_sapiens.GRCh38.87.exon.bed
awk '$6 == "+"' Homo_sapiens.GRCh38.87.exon.bed | awk -F"\t" '!seen[$1, $2]++' | grep -v '\.1' > Homo_sapiens.GRCh38.87.exon.plus.bed
awk '$6 == "-"' Homo_sapiens.GRCh38.87.exon.bed | awk -F"\t" '!seen[$1, $3]++' | grep -v '\.1' | grep -v '\.2' > Homo_sapiens.GRCh38.87.exon.minus.bed
exFile=Homo_sapiens.GRCh38.87.exon.plus.bed
awk '{$2 = $2 - 21; print}' $exFile | awk '{OFS="\t";} {$3 = $2 + 20; print}' | fastaFromBed -fi ~/DNase_ENCODE/hg38.fa -s -bed stdin -fo exon.hg38.20.fasta
declare -a arr=("cag" "tag" "gag" "aag")
for i in "${arr[@]}"
do
echo $i
grep -B 1 -i $i exon.hg38.fasta | grep '>' > exon.${i}.hg38.fasta
cat exon.${i}.hg38.fasta | awk -F">" '{print $NF}' | awk -F":" '{print $1}' > exon.${i}.chr.txt
cat exon.${i}.hg38.fasta | awk -F":" '{print $NF}' | awk -F"-" '{print $1}' > exon.${i}.start.txt
cat exon.${i}.hg38.fasta | awk -F"-" '{print $NF}' | awk -F"(" '{print $1}' > exon.${i}.end.txt
cat exon.${i}.hg38.fasta | awk -F"(" '{print $NF}' | awk -F")" '{print $1}' > exon.${i}.strand.txt
paste exon.${i}.chr.txt exon.${i}.start.txt exon.${i}.end.txt exon.${i}.strand.txt exon.${i}.strand.txt exon.${i}.strand.txt > exon.${i}.hg38.bed
fastaFromBed -fi ~/DNase_ENCODE/hg38.fa -s -bed exon.${i}.hg38.bed -fo exon.${i}.hg38.3nuc.fasta
rm exon.*.hg38.*.txt
done
source('https://raw.githubusercontent.com/guertinlab/seqOutBias/master/docs/R/seqOutBias_functions.R')
exon.plus = read.table('~/Core_PRO/Homo_sapiens.GRCh38.87.exon.plus.bed')
exon.plus[,3] = exon.plus[,2] + 149
exon.plus[,2] = exon.plus[,2] - 151
exon.minus = read.table('~/Core_PRO/Homo_sapiens.GRCh38.87.exon.minus.bed')
exon.minus[,2] = exon.minus[,3] - 150
exon.minus[,3] = exon.minus[,3] + 150
plus.exon.pro = composites.test.naked('/Users/guertinlab/Core_PRO/plus', exon.plus, region = 150, grp = 'PRO-seq')
plus.exon.pro[[1]]$x = plus.exon.pro[[1]]$x -0.5
composites.func.panels.naked.chromatin(plus.exon.pro[[1]], fact = 'PolII', summit = 'Exon plus', num=30)
minus.exon.pro = composites.test.naked('/Users/guertinlab/Core_PRO/minus', exon.minus, region = 150, grp = 'PRO-seq')
minus.exon.pro[[1]]$x = minus.exon.pro[[1]]$x -0.5
composites.func.panels.naked.chromatin(minus.exon.pro[[1]], fact = 'PolII', summit = 'Exon minus -', num=30)
save(minus.exon.pro, plus.exon.pro, file = '~/Core_PRO/exon.pro.Rdata')
load('~/Core_PRO/exon.pro.Rdata')
#acceptor seqLogo
exonjunc = read.table('exon.hg38.20.fasta', comment.char = '>') exonjunc[,1] = as.character(exonjunc[,1])
exonjunc = data.frame(lapply(exonjunc, function(v) {
if (is.character(v)) return(toupper(v))
else return(v)
}))
pswm.func(exonjunc[,1], 'splice_acceptor', positions = 20)
#subdividing sequences at acceptor site
exon.aag = read.table('~/Core_PRO/exon.aag.hg38.bed')
exon.cag = read.table('~/Core_PRO/exon.cag.hg38.bed')
exon.gag = read.table('~/Core_PRO/exon.gag.hg38.bed')
exon.tag = read.table('~/Core_PRO/exon.tag.hg38.bed')
#selecting the same number of coordinates to generate coparable confidence interval estimates
exon.cag = randomRows(exon.cag, nrow(exon.tag))
exon.cag[,3] = exon.cag[,2] + 153
exon.cag[,2] = exon.cag[,2] - 147
plus.exon.cag.pro = composites.test.naked('/Users/guertinlab/Core_PRO/plus', exon.cag, region = 150, grp = 'PRO-seq')
plus.exon.cag.pro[[1]]$x = plus.exon.cag.pro[[1]]$x -0.5 composites.func.panels.naked.chromatin(plus.exon.cag.pro[[1]], fact = 'PolII', summit = 'Exon CAG plus', num=30)
exon.tag[,3] = exon.tag[,2] + 153
exon.tag[,2] = exon.tag[,2] - 147
plus.exon.tag.pro = composites.test.naked('/Users/guertinlab/Core_PRO/plus', exon.tag, region = 150, grp = 'PRO-seq')
plus.exon.tag.pro[[1]]$x = plus.exon.tag.pro[[1]]$x -0.5 composites.func.panels.naked.chromatin(plus.exon.tag.pro[[1]], fact = 'PolII', summit = 'Exon TAG plus', num=30)
exon.aag[,3] = exon.aag[,2] + 153
exon.aag[,2] = exon.aag[,2] - 147
plus.exon.aag.pro = composites.test.naked('/Users/guertinlab/Core_PRO/plus', exon.aag, region = 150, grp = 'PRO-seq')
plus.exon.aag.pro[[1]]$x = plus.exon.aag.pro[[1]]$x -0.5 composites.func.panels.naked.chromatin(plus.exon.aag.pro[[1]], fact = 'PolII', summit = 'Exon AAG plus', num=30)
save(minus.exon.pro, plus.exon.pro, plus.exon.aag.pro, plus.exon.cag.pro, plus.exon.tag.pro, file = '~/Core_PRO/exon.pro.Rdata')
load('~/Core_PRO/exon.pro.Rdata')


seqOutBias.
seqOutBias.
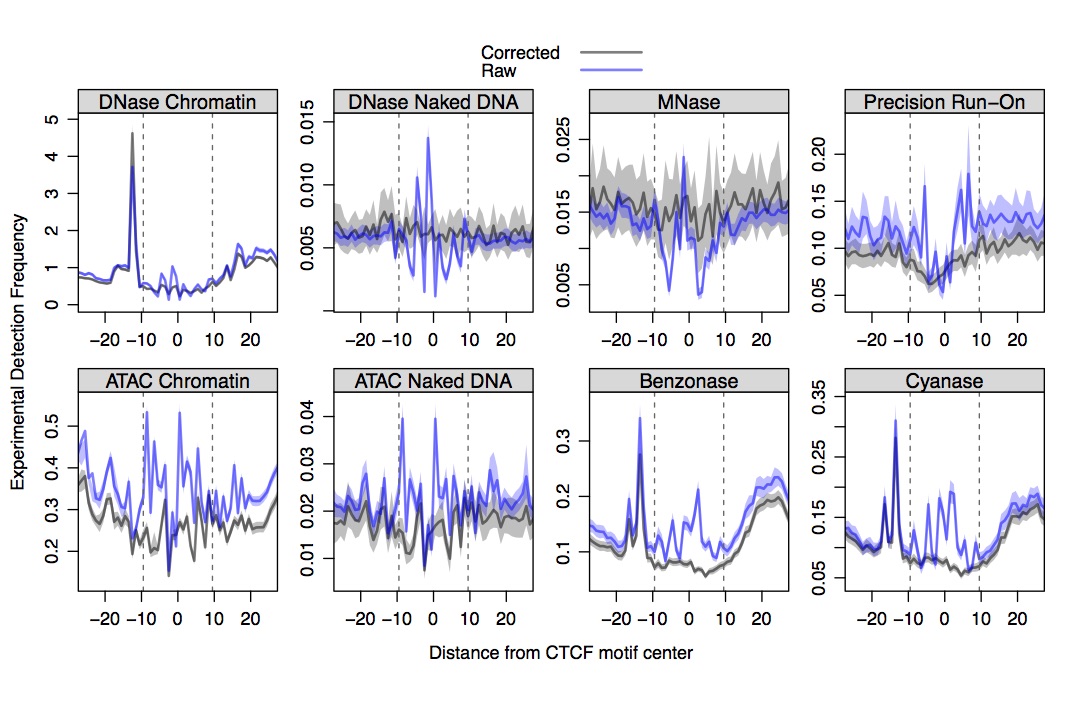
Plotting seqOutBias correction of DNase, MNase, ATAC, TACh, and PRO-seq data at CTCF binding sites.
The only factor with ChIP-seq data in MCF7, GM12878, K562, and mouse liver is CTCF. SeqOutBias corrects the sequence bias for CTCF reasonably well. Although, the Tn5 bias seems to span a wide domain and a k-mer correction is likely not optimal, as sequence features that span this domain likely influence Tn5 recognition.
source('https://raw.githubusercontent.com/guertinlab/seqOutBias/master/docs/R/seqOutBias_functions.R')
load('~/DNase_ENCODE/MCF7_composites.Rdata')
load('~/TACh_Grontved/MOUSE_composites.Rdata')
load('~/MNase_Zhang/all.composites.mnase.mcf7.Rdata')
load('~/ATAC_Walavalkar/ATAC_naked_composites.Rdata')
load('~/Core_PRO/PRO_composites.Rdata')
#Comparing correction of sequence bias dictated by the CTCF motif
#all.composites.ATAC$grp = gsub("CTCF_GM12878", "CTCF", all.composites.ATAC$grp)
all.composites.ATAC$cond = gsub("ATAC_GM12878_no_scale_merged", "ATACgm_0-mer", all.composites.ATAC$cond)
all.composites.ATAC$cond = gsub("ATAC_GM12878_NXNXXXCXXNNXNNNXXN_NXXNNNXNNXXCXXXNXN_merged", "ATACgm_NXNXXXCXXNNXNNNXXN-mer",
all.composites.ATAC$cond)
all.composites.ATAC.naked$cond = gsub("C1_gDNA_no_scale_merged", "ATACnk_0-mer", all.composites.ATAC.naked$cond)
all.composites.ATAC.naked$cond = gsub("C1_gDNA_NXNXXXCXXNNXNNNXXN_NXXNNNXNNXXCXXXNXN_merged", "ATACnk_NXNXXXCXXNNXNNNXXN-mer",
all.composites.ATAC.naked$cond)
all.composites.plus.pro$cond = gsub("PRO_plus_body_0-mer", "PRO_0-mer", all.composites.plus.pro$cond)
all.composites.plus.pro$cond = gsub("PRO_plus_body_NNNCNNN-mer", "PRO_6-mer", all.composites.plus.pro$cond)
alldf = rbind(all.composites.cyanase, all.composites.benzonase, all.composites.dnase.mcf7, all.composites.dnase.naked, all.composites.ATAC, all.composites.ATAC.naked, all.composites.mnase.mcf7, all.composites.plus.pro)
#colnames(alldf) = c('est', 'x', 'grp', 'upper', 'lower', 'cond')
ctcf.df = alldf[alldf$grp == 'CTCF',]
ctcf.df = ctcf.df[ctcf.df$cond == 'Cyanase_0-mer' | ctcf.df$cond == 'Cyanase_10-mer' | ctcf.df$cond == 'Benzonase_0-mer' |
ctcf.df$cond == 'Benzonase_10-mer' | ctcf.df$cond == 'MCF7_0-mer' | ctcf.df$cond == 'MCF7_6-mer' | ctcf.df$cond == 'Naked_0-mer' | ctcf.df$cond == 'Naked_6-mer' | ctcf.df$cond == 'ATACgm_0-mer' |
ctcf.df$cond == 'ATACgm_NXNXXXCXXNNXNNNXXN-mer' | ctcf.df$cond == 'ATACnk_0-mer' | ctcf.df$cond == 'ATACnk_NXNXXXCXXNNXNNNXXN-mer' | ctcf.df$cond == 'MNase_0-mer' |
ctcf.df$cond == 'MNase_8-mer' | ctcf.df$cond == 'PRO_0-mer' | ctcf.df$cond == 'PRO_6-mer',]
ctcf.df$grp = sapply(strsplit(as.character(ctcf.df$cond),'_'), "[", 1)
ctcf.df$cond = sapply(strsplit(as.character(ctcf.df$cond),'_'), "[", 2)
ctcf.df[ctcf.df=="ATACgm"] = 'ATAC Chromatin'
ctcf.df[ctcf.df=="MCF7"] = 'DNase Chromatin'
#ctcf.df[ctcf.df=="MNase"] = 'DNase Chromatin'
ctcf.df[ctcf.df=="Naked"] = 'DNase Naked DNA'
ctcf.df[ctcf.df=="ATACnk"] = 'ATAC Naked DNA'
ctcf.df[ctcf.df=="ATACnk"] = 'ATAC Naked DNA'
ctcf.df[ctcf.df=="PRO"] = 'Precision Run-On'
ctcf.df[ctcf.df=='0-mer'] = 'raw'
ctcf.df[ctcf.df=="10-mer"] = 'corrected'
ctcf.df[ctcf.df=="6-mer"] = 'corrected'
ctcf.df[ctcf.df=="8-mer"] = 'corrected'
ctcf.df[ctcf.df=="NNNXXNCNN-mer"] = 'corrected'
composites.func(ctcf.df, fact= "Experimental", summit= "CTCF motif",num = 24,
col.lines = rev(c(rgb(0,0,1,1/2), rgb(0,0,0,1/2))),
fill.poly = rev(c(rgb(0,0,1,1/4), rgb(0,0,0,1/4))))